Jake Yeung
Theory, Experiment, and Analysis for Genome Biology and Drug Discovery

Principal Scientist.
Genentech, Inc.
1 DNA Way,
South San Francisco, CA 94080, USA
Systems Biology for Drug Discovery
I am a systems biologist at Genentech’s Research & Early Development (gRED), where I combine theory, experiment, and analysis to understand how cells regulate their genomes and to discover drugs that revert disease states to healthy ones. My work pairs top-down observational analysis of complex tissue environments with bottom-up causal perturbations to isolate specific cellular behaviors. By integrating public and in-house datasets, I aim to predict therapeutic efficacy and potential off-target effects across diverse patient contexts.
For my training, I was a Human Frontier Science Program (HFSP) fellow with Alexander van Oudenaarden at the Hubrecht Institute, and completed my PhD at EPFL in computational systems biology advised by Felix Naef.
Research themes
My projects span three approaches — theory, experiment, and statistical analysis — and typically pair two of these at a time (Fig. 1), bringing in both wet and dry lab components to address questions in oncology and genome biology.
(1) Theory-driven experimental design
Theory ↔ Experiment.
Designing informative experiments requires theory: models that predict what can be learned from a given measurement strategy before running it. Rather than experimentally enumerating all possible conditions, we derive theoretical frameworks that quantify information gain, reproducibility limits, and cost-information tradeoffs, then use these to design experiments that maximize signal per dollar.
We have developed an experimental, computational, and theoretical framework to evaluate cost-efficient Perturb-seq designs (preprint: Yeung et al 2026).
We have also developed a statistical unmixing method (scChIX-seq) that maps multiple histone modifications in single cells and learns the correlation structure between histone marks (Yeung*, Florescu*, Zeller* et al 2023).
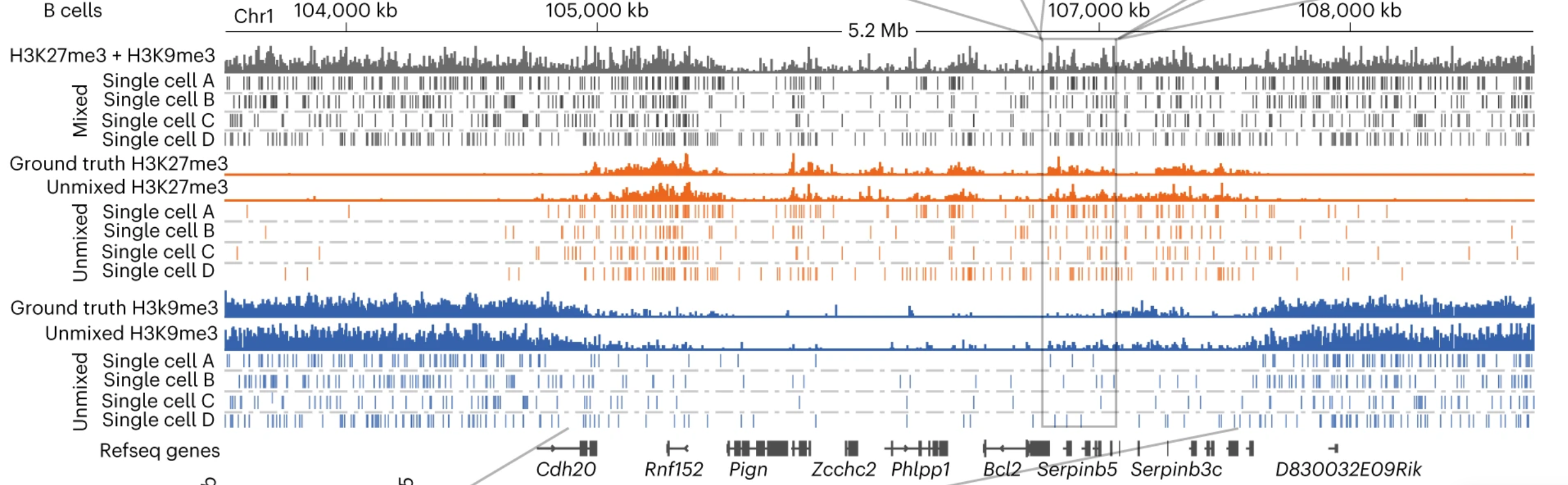
(2) Statistical analysis of functional genomics technologies
Experiment ↔ Statistical Analysis.
New functional genomics technologies can measure the regulation and output of every gene across virtually every cell in the body. Making sense of this data requires statistical methods that separate biological signal from technical artifacts in sparse, noisy settings, and that integrate information across experiments performed under vastly different conditions. The goal is inference and prediction that can guide the next experiment or therapeutic hypothesis.
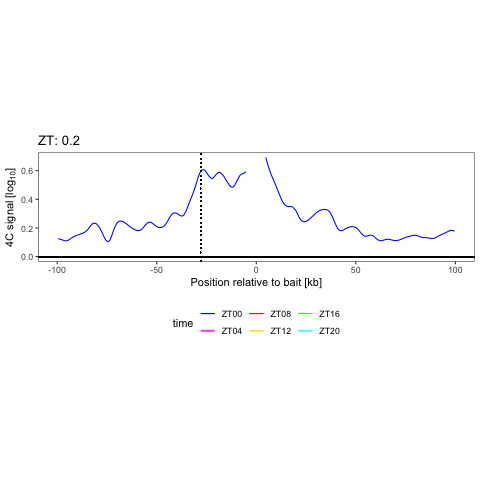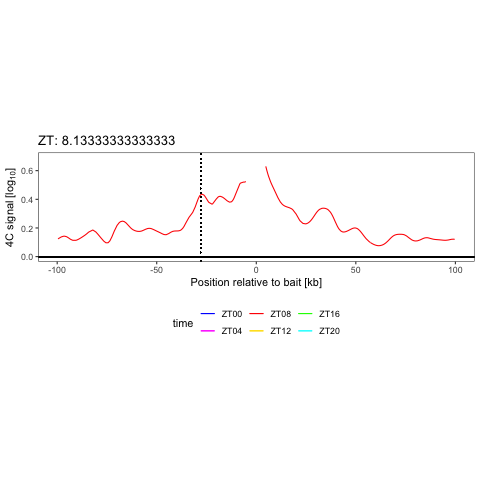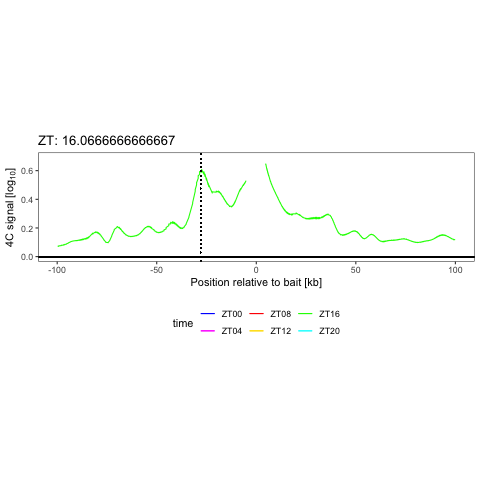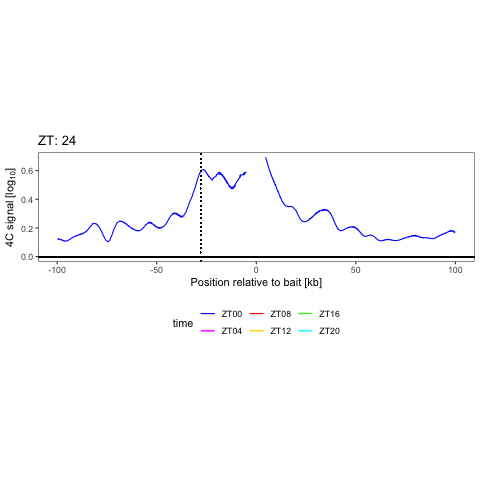
We have shown that combining cell surface markers with histone modification mapping in single cells (sortChIC) can systematically compare genome-wide differences between active (e.g. H3K4me1, H3K4me3) and repressed (e.g. H3K27me3, H3K9me3) chromatin states during blood maturation (Zeller*, Yeung* et al 2022). At Genentech, we extend these approaches to large-scale perturbation screens in oncology, integrating in-house and public datasets to predict therapeutic response across patient contexts.
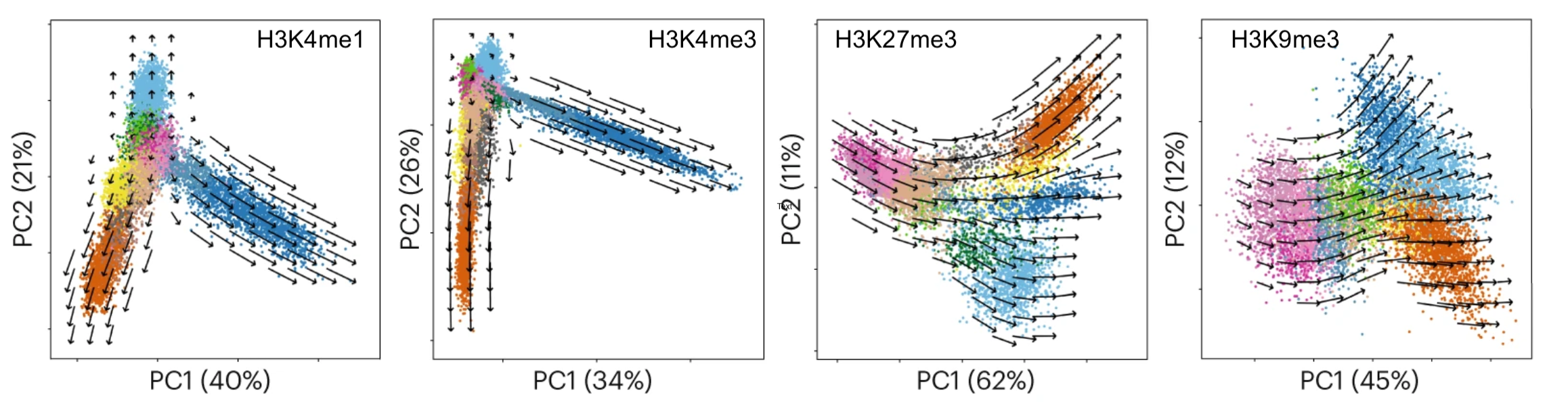
(3) Principles of genome regulation
Theory ↔ Statistical Analysis.
Understanding how cells regulate their genomes requires moving from description to mechanism: hypothesizing regulatory principles, articulating them as statistical models, and testing them against carefully crafted null hypotheses. Even well-characterized tissues harbor rich regulatory structure that only becomes visible when the right question is asked of the right data.
We have developed model selection approaches to categorize genes by shared regulatory logic, reducing diverse expression patterns across a tissue to a small number of interpretable principles — for example, quantifying how much tissue-specific dynamics come from individual transcription factors versus cooperation between pairs (Yeung*, Mermet* et al 2018), or disentangling sleep-wake driven from circadian processes in gene expression (Hor*, Yeung* et al 2019). These analyses yield direct predictions testable by experiment, such as CRISPR knockout of enhancers (Mermet*, Yeung* et al 2018), and provide a template for the same regulatory decomposition in disease contexts.